揭示机器学习中化学键的秘密
一种新的机器学习方法提供了对催化的重要见解,催化是一个基本过程,可以减少有毒废气的排放或生产诸如织物之类的重要材料。在《自然通讯》上发表的一份报告中,弗吉尼亚理工大学化学工程副教授辛宏亮及其研究人员开发了一种化学吸附的贝叶斯学习模型,简称Bayeschem,旨在利用人工智能来解锁化学键的本质。在催化剂表面。
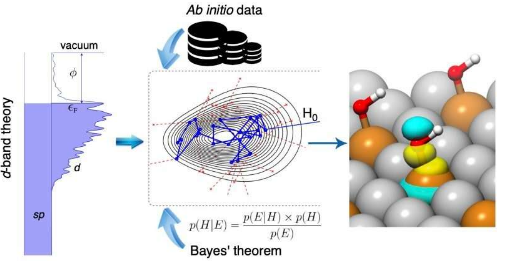
辛说:“一切都取决于催化剂如何与分子结合。” “相互作用必须足够强,以在合理的低温下破坏某些化学键,但又不能太强,以至于催化剂会被反应中间体中毒。这一规则被称为催化中的萨巴帖原理。”
辛说,了解催化剂如何与不同的中间体相互作用,并确定如何控制它们的结合强度,使它们处于“金锁区域”内,这是设计有效催化过程的关键。该研究为此目的提供了一种工具。
Bayeschem使用贝叶斯学习来工作,贝叶斯学习是一种用于从数据推断模型的特定机器学习算法。“假设您有一个基于完善的物理定律的领域模型,并且您想用它来进行预测或学习有关世界的新知识,”前化学工程博士学位的学生Siwen Wang解释说。“贝叶斯方法是根据我们的先验知识和观察到的数据(通常是稀缺的)来学习模型参数的分布,同时提供模型预测的不确定性量化。”
Bayeschem中使用的d带化学吸附理论是一种描述在固体表面上化学键合的理论,其中涉及的d电子通常形状像四叶草。该模型解释了催化剂原子的d轨道是如何重叠并吸引到具有球形或哑铃状形状的吸附价原子轨道上的。它一直被认为是标准模型中的多相催化自20世纪90年代开发的锤子和Nørskov,虽然它已经成功地解释许多系统的结合趋势,鑫说,模型有时失败,因为电子相互作用的内在复杂性。
据Xin说,Bayeschem将d波段理论带入了一个新的水平,以量化那些相互作用的强度,并可能定制一些旋钮,例如结构和成分,以设计更好的材料。该方法通过扩展其吸附性能的预测和解释能力来推进化学吸附的d波段理论,这两者在催化剂发现中都至关重要。但是,与通过大量数据训练的黑匣子机器学习模型相比,Bayeschem的预测准确性仍然可以改善,Xin小组的化学工程博士生Hemanth Pillai表示,他同样为这项研究做出了贡献。
辛说:“有机会提出基于深度学习算法和化学吸附理论的高度准确和可解释的模型,这对于实现催化中人工智能的目标是非常有益的。”
标签: 机器学习