人工智能模型发现潜在药物分子的速度要快一千倍
几何深度学习模型比最先进的计算模型更快、更准确,从而降低了药物试验失败的机会和成本。整个已知宇宙都充满了无数的分子。但是这些分子中有多少具有潜在的类似药物的特征,可用于开发挽救生命的药物治疗?百万?数十亿?万亿?
答案是:novemdecillion,或1060。这个庞大的数字延长了Covid-19等快速传播疾病的药物开发过程,因为它远远超出了现有药物设计模型可以计算的范围。换个角度来看,银河系大约有1亿颗或108颗恒星。
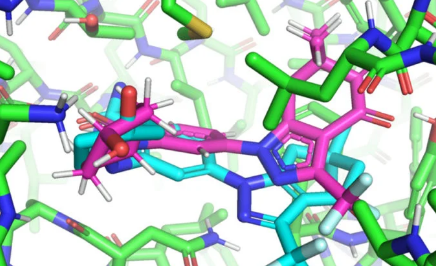
在将在国际机器学习会议(ICML)上发表的一篇论文中,麻省理工学院的研究人员开发了一种名为EquiBind的几何深度学习模型,它比现有最快的计算分子对接模型QuickVina2-W快1200倍,成功地将药物样分子与蛋白质结合。
EquiBind基于其前身EquiDock,后者专门使用已故的Octavian-EugenGanea开发的技术结合两种蛋白质,该技术由最近的麻省理工学院计算机科学和人工智能实验室和AbdulLatifJameelClinicforMachineLearninginHealth(JameelClinic)博士后,他也是EquiBind论文的合著者。
在药物开发开始之前,药物研究人员必须找到有希望的药物样分子,这些分子可以在称为药物发现的过程中正确结合或“对接”到某些蛋白质靶标上。在成功与蛋白质对接后,结合药物(也称为配体)可以阻止蛋白质发挥作用。如果这种情况发生在细菌的一种必需蛋白质上,它可以杀死细菌,从而保护人体。
然而,药物发现的过程在财务和计算上都可能是昂贵的,数十亿美元投入到这个过程中,并且在食品和药物管理局最终批准之前进行了十多年的开发和测试。更重要的是,90%的药物在人体试验后由于没有效果或副作用过多而失败。制药公司收回这些失败成本的方法之一是提高成功药物的价格。
当前寻找有希望的候选药物分子的计算过程是这样的:大多数最先进的计算模型依赖于大量候选样本以及评分、排名和微调等方法,以获得最佳的“拟合”。配体和蛋白质。
HannesStärk是麻省理工学院电气工程和计算机科学系的一年级研究生,也是该论文的主要作者,他将典型的配体与蛋白质结合方法比作“试图将钥匙插入有很多锁孔的锁中”。”在选择最佳模型之前,典型模型会花费大量时间对每个“拟合”进行评分。相比之下,EquiBind只需一步即可直接预测精确的关键位置,而无需事先了解蛋白质的目标口袋,这被称为“盲对接”。
与大多数需要多次尝试才能找到配体在蛋白质中的有利位置的模型不同,EquiBind已经具有内置的几何推理功能,可帮助模型学习分子的基本物理特性,并在遇到新的、看不见的数据时成功泛化以做出更好的预测.
这些发现的发布很快引起了行业专业人士的关注,其中包括RelayTherapeutics的首席数据官PatWalters。沃尔特斯建议该团队在已经存在的用于肺癌、白血病和胃肠道肿瘤的药物和蛋白质上尝试他们的模型。尽管大多数传统的对接方法未能成功地结合对这些蛋白质起作用的配体,但EquiBind成功了。
“EquiBind为对接问题提供了一种独特的解决方案,它结合了姿势预测和结合位点识别,”Walters说。“这种方法利用了来自数千个公开可用的晶体结构的信息,有可能以新的方式影响该领域。”
“我们很惊讶,虽然所有其他方法都完全错误或只有一个正确,但EquiBind能够将其放入正确的口袋中,因此我们很高兴看到这一结果,”Stärk说。
虽然EquiBind收到了来自行业专业人士的大量反馈,这些反馈帮助团队考虑了计算模型的实际用途,但Stärk希望在7月即将举行的ICML上找到不同的观点。
“我最期待的反馈是关于如何进一步改进模型的建议,”他说。“我想与那些研究人员讨论……告诉他们我认为下一步可以做什么,并鼓励他们继续前进,将模型用于他们自己的论文和他们自己的方法……我们已经有许多研究人员已经伸出援手,并询问我们是否认为该模型对他们的问题有用。”
这项工作部分由药物发现和合成联盟资助;贾米尔诊所;DTRA发现针对新出现的威胁的医疗对策计划;DARPA加速分子发现计划;麻省理工学院-武田奖学金;和NSFExpeditions授予协作研究:通过代码了解世界。
这部作品是为了纪念Octavian-EugenGanea,他为几何机器学习研究做出了重要贡献,并慷慨地指导了许多学生——一位有着谦逊灵魂的杰出学者。
标签: